Proteomics
|
|
|
|
 |
|
Proteine sind äußerst vielseitige Makromoleküle, was man ihnen deutlich ansieht
|
Die Gesamtheit der exprimierten Proteine einer Zelle bezeichnet man als Proteom - in Analogie zum Genom als der Gesamtheit des Erbguts. Zwischen Genom und Proteom bestehen in der Tat große Unterschiede was die Art der Informationen betrifft. Zum Beispiel gibt es nur schwache Korrelationen von mRNA- und Protein-Expressionsmustern. Quantitative Änderungen der mRNA-Transkription korrelieren allenfalls zu 50% mit der Proteinbildung, was bedeutet, dass Analysen auf der RNA-Ebene nur unvollständig Auskunft über zelluläre Prozesse geben. Auch besteht nur selten ein 1:1-Verhältnis von Genen und Proteinen. Bei der Hefe etwa, gibt es zu jedem Gen im Durchschnitt drei veschiedene Proteinprodukte. Das kann daran liegen, dass mehrere Proteine innerhalb eines Start- und Stoppsignals codiert sind oder dass aus einer exprimierten Proteinkette durch nachträgliche Spaltung mehrere funktionale Proteine entstehen, was man an der zugehörigen DNA-Sequenz nicht leicht erkennen kann. Zudem wird bei höheren Organismen die Funktion vieler Proteine durch enzymatische Veränderungen nach der Translation entscheidend beeinflußt. Beim Menschen schätzt man bis zu zehn unterscheidbare, jeweils anders modifizierte Proteine pro Gen, d.h. etwa 300.000 verschiedene Proteine.
Unter dem Schlagwort Proteomics (Proteomforschung) fasst man Forschungsprojekte zusammen, die darauf gerichtet sind, das Proteinrepertoire zu erfassen, die Funktionen der Proteine zu entschlüsseln und Wechselwirkungen zwischen den einzelnen Proteinen zu entwirren. Proteomics erfordern eine Vielzahl an Techniken, die Zusammenarbeit von Wissenschaftlern unterschiedlichster Disziplinen, großen apparativen Aufwand und enorme Computerleistung. Drei klar unterscheidbare Arbeitsgebiete der Proteomforschung sollen nachfolgend kurz vorgestellt werden:
|
Analyse der Proteinexpression. Mit Hilfe von Trennverfahren wie der Gelelektrophorese kann man die Zahl, Größe und chemische Natur der exprimierten Proteine eines Zellzustands erfassen. Eine unverzichtbare Hilfe bei der Charakterisierung der Proteine ist die Massenspektrometrie geworden. Hier genügt es, wenn bereits kleinste Mengen der Peptidfragmente eines isolierten Proteins zur Verfügung stehen, um Sequenzinformationen zu gewinnen. Durch den Vergleich der Proteinexpressionsmuster von verschiedenen Zellzuständen erfahren die Wissenschaftler, welche Eiweisse in welchem Zustand vorhanden sind und in welchem sie fehlen. Für die Pharmaforschung sind solche Hinweise sehr wertvoll geworden: Wenn man z.B. die Eiweisse kennt, die nach der Zugabe eines Wirkstoffs von der Zelle gebildet werden, kann man herausfinden, wodurch die Wirkung zustandekommt und mit diesen Informationen möglicherweise bessere Wirkstoffe entwickeln.
|

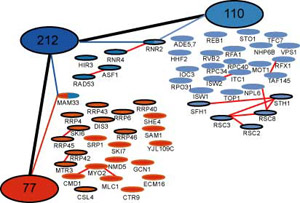
Protein Mapping Die Inventarisierung der Proteine einer Zelle bzw. eines Zellzustands und die Kenntnis ihrer Sequenzen liefert nur selten Informationen über ihre Funktionen. Hier muß man z.B. auch berücksichtigen, dass viele Proteine aus Untereinheiten zusammengesetzt sind und nur als Komplexe eine biologische Funktion ausüben und dass die meisten zellbiologischen Phänomene durch das Zusammenspiel von Proteinen zustandekommen. Beste Beispiele hierfür sind zahlreiche Signalleitungsprozesse, die externe molekulare Signale auf der äußeren Zellmembranoberfläche über Rezeptorproteine aufnehmen und über einige nachfolgend "geschaltete" Proteinkaskade im Zellinneren bis in den Zellkern leiten, wo z.B. Gene aktiviert oder abgeschaltet werden. Das Zusammenspiel der Signalproteine erfordert zwangsläufig ihren physikalischen Kontakt. Um diese Wechselwirkungen zu untersuchen, wurden leistungsfähige Methoden entwickelt. Mit ihnen kann man die Interaktionen innerhalb großer Proteinpools feststellen. Als Grafik aufgetragen zeigen sie ein wirres Geflecht von bilateralen Wechselwirkungen (protein mapping, map= Karte). Aus dem mapping können sich wertvolle Auskünfte zur Funktion eines Proteins ergeben, da mitunter bereits bekannte Proteine als Partner auftauchen. Besonders wertvolle Informationsquellen sind evolutionäre Verwandtschaften zwischen Proteinen aus unterschiedlichen Organismen. Dabei findet man z.B., daß zwei Domänen, die bei einem Organismus innnerhalb eines Proteins auftauchen, beim anderen Organismus aber auf zwei Proteine verteilt sind. In solchen Fällen bestätigt sich oft, dass die separaten Proteine tatsächlich über die beiden Domänen miteinander wechselwirken. |
Strukturforschung (structural genomics) Das mapping liefert im Idealfall deutliche Hinweise auf die Affinität eines Proteins zu einem anderen Eiweiss und damit auch auf seine Funktion. Um jedoch zu verstehen, welche Partien des mitunter riesigen Proteins für den Effekt verantwortlich sind, oder bevor man - bei pharmazeutisch interessanten Kandidaten - mit passenden Effektormolekülen auf das Zusammenspiel Einfluß ausüben kann, muß man die molekulare Architektur des Moleküls herausfinden. Das ist nicht so leicht. Proteine sind große, meistens sehr empfindliche Moleküle, mitunter aus vielen Untereinheiten zusammengesetzt und in vielen Fällen auch mittels Gentechnik nur schwer in der benötigten Menge und Reinheit zu gewinnen. Die Aufklärung der molekularen Strukturen mit physikalischen Meßmethoden wie Röntgenbeugung und Kernspinresonanz NMR (s. Kasten) ist ebenfalls nicht trivial und meistens zeitaufwändig. Bei all diesen Teilschritten wurden in den letzten Jahren jedoch große methodische Fortschritte in der Automatisierung gemacht, so daß man die "Massenproduktion" von gelösten Proteinstrukturen bereits unter dem Stichwort Structural Genomics zusammenfasst. |
Neue Herausforderungen: Biologische Systeme
Zunehmend wird klar, dass die Bearbeitung auch von zahlreichen einzelnen Stoffwechsel-
oder Signalleitungswegen uns nicht zwangsläufig dem Verständnis der
Funktion eines gesamten zellulären Systems näherbringt. Denn viele
neue Fragen stellen sich dabei: Wie interagieren verschiedene Teilsysteme, z.B.
Signalleitungsketten? Wie antwortet das gesamte System auf äußere
Störungen? Wie werden komplexe Signale erkannt und interpretiert? Um diese
schwierigen Fragen anzugehen, muß man die Forschung vieler Arbeitsgruppen
koordinieren und fokussieren und das know how ganz unterschiedlicher Forschungsrichtungen
zusammenführen. Die Herausforderung, die Netzwerke biologischer Systeme
vollkommen verstehen zu wollen, führt zunächst erstmal zu Netzwerken
aus vielen Forschungslabors. Auf diesem noch sehr jungen, als Systembiologie
bezeichneten Forschungsbebiet werden Zellbiologen, Regelungstechniker, Biochemiker,
Informatiker und Physikochemiker noch viele Jahre eng zusammenarbeiten müssen
bis einmal ein komplettes biologisches System - z.B. eine Zelle - als dichtes
Netzwerk zahlloser funktioneller Einheiten in einem Computer abgebildet werden
kann. Für das Verständnis lebender Systeme ließe sich aus solchen
Modellen viel lernen. Auch praktische Anwendungen liegen auf der Hand: So könnten
in der Zukunft viele Experimente in silico
ausgeführt werden. Realistisch konzipierte virtuelle Zellen würden
viele Fragen beantworten, vielleicht sogar die Entwicklung von Arzneimitteln
erleichtern und zugleich die Zahl der Tierversuche verringern. Aber bis dahin
wird es wahrscheinlich noch einige Jahre dauern; die Systembiologie steht gerade
erst am Anfang.
